The agency requests additional manufacturing information and does not cite safety or efficacy concerns or require new clinical studies.
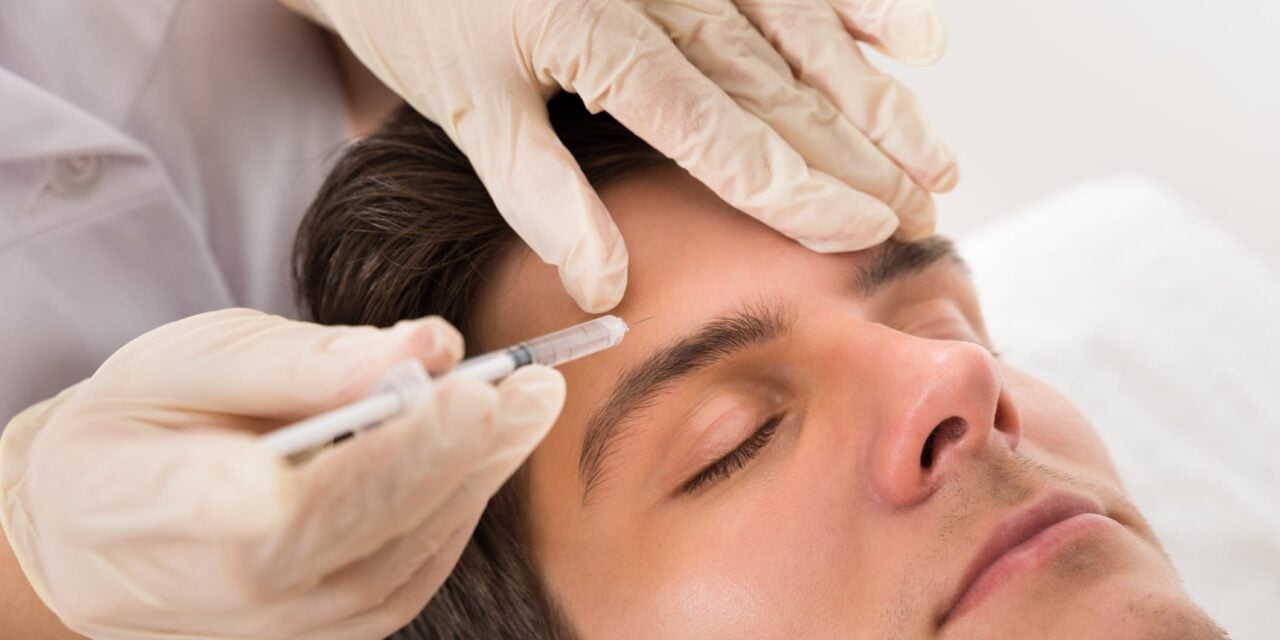
AbbVie announced it received a Complete Response Letter from the US Food and Drug Administration regarding its Biologics License Application for trenibotulinumtoxinE (TrenibotE), a first-in-class botulinum neurotoxin serotype E characterized by a rapid onset of action as early as eight hours after administration and short duration of effect of two to three weeks.
According to the company, the FDA’s letter requests additional information related to manufacturing processes. The agency did not identify safety or efficacy concerns and did not request additional clinical studies.
AbbVie says it plans to address the FDA’s comments and submit a response in the coming months.
“We strongly believe trenibotulinumtoxinE is an important innovation in botulinum toxin science, with the potential to expand options for patients interested in facial aesthetics,” says Roopal Thakkar, executive vice president, research and development, and chief scientific officer at AbbVie, in a release. “Though disappointed, we remain confident in the strength and integrity of our application, and we are well-positioned to respond to the agency’s feedback promptly to support completion of the review.”
Regulatory reviews for TrenibotE are ongoing in other countries, according to the company.
TrenibotE is a botulinum neurotoxin serotype E under investigation for aesthetic use, including the treatment of moderate to severe glabellar lines. AbbVie reports that its clinical program included more than 2,100 patients across multiple Phase 3 studies.
ID 57712784 © Andrey Popov | Dreamstime.com



